COSG是什么
COSG(COSine similarity-based marker Gene identification)是由来自哈佛医学院和Broad研究所博后Ming Dai等人开发,旨在从余弦相似度的角度鉴定cluster的marker gene,于2021年12月被Briefings in Bioinformatics接收。目前已将此方法分别包装为R包与Python包,分别对应Seurat与Scanny分析流程。
为什么选择COSG
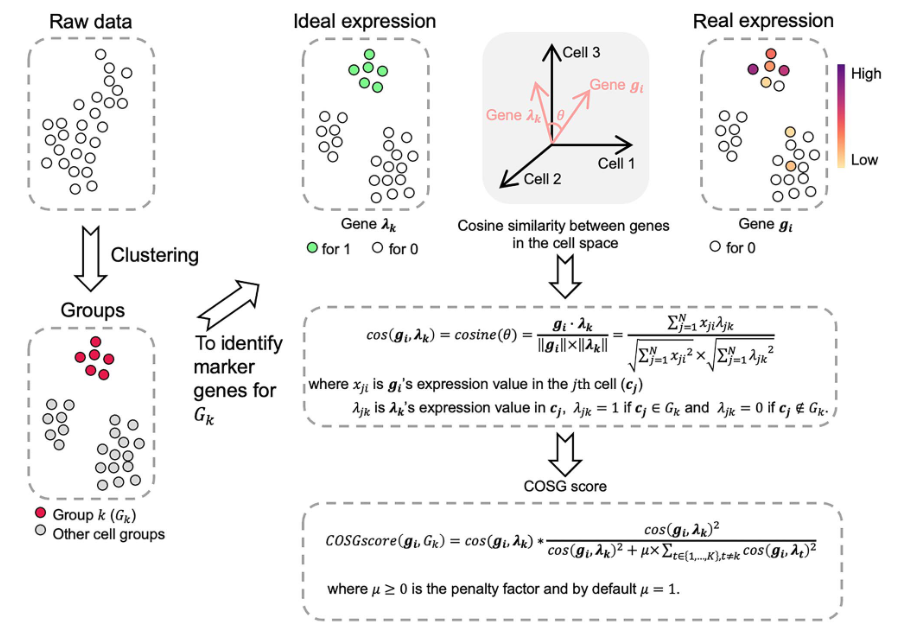
传统的marker基因鉴定方法基于欧式距离,即目标细胞类群与其它细胞之间的基因表达差异来进行marker基因的鉴定。但这样的鉴定方法存在潜在的问题:当marker基因在目标细胞类群以外的其它一小部分细胞中高表达时(如某个小的细胞亚型),传统的鉴定方法仍然会将其鉴定为marker基因,但因为小范围的高表达对于整体的细胞群来说不具备决定性意义,所以仍然会认为这个marker基因在其余的细胞群中是低表达的。而COSG(COSine similarity-based marker Gene identification),即基于余弦值的marker基因鉴定方法可以解决上述问题。原因在于余弦值不依赖于向量的模,放到单细胞分析的背景下就是不依赖于基因的表达量,而依赖于基因的表达模式。举例为证:基因A在细胞1和2中的表达量分别是2和4,基因B在细胞1和2中的表达量分别是1和2,如果根据欧式距离的算法,显然基因A和基因B会被定义成为不同的表达模式,但是如果我们基于余弦值的话,两个基因在该细胞集合中的表达向量之间的夹角就为:
在这个背景下,两个基因表达向量之间的夹角为0,也就是这两个基因在这个细胞集合中的表达模式实际上是相同的。
怎么做COSG分析
COSG分析思路
(1)准备好数据, 即标准化后的单细胞表达矩阵,并完成细胞聚类分析(scATAC, ST同理);
(2)对cluster K模拟一个ideal marker gene(仅在k表达,其余cluster不表达);
(3)计算所有真实基因分别与该基因的余弦相似度,并进一步计算出COSG score;
(4)COSG score取值范围为0~1。值越大,表示为cluster specific marker gene;
(5)同理依次计算出其余cluster的marker gene。
与其他方法的性能比较
文章主要与常用的Wilcoxon-test等分析方式在多种数据集上进行比较,概括如下:
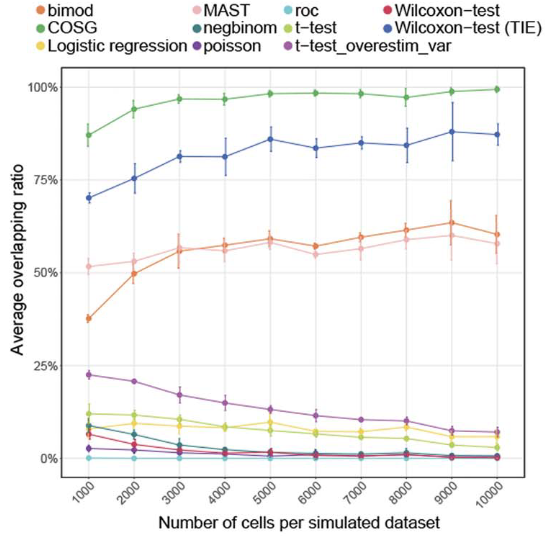
(1)在模拟的单细胞数据集中,COSG方法可以最大程度发现每个clutser的marker gene;
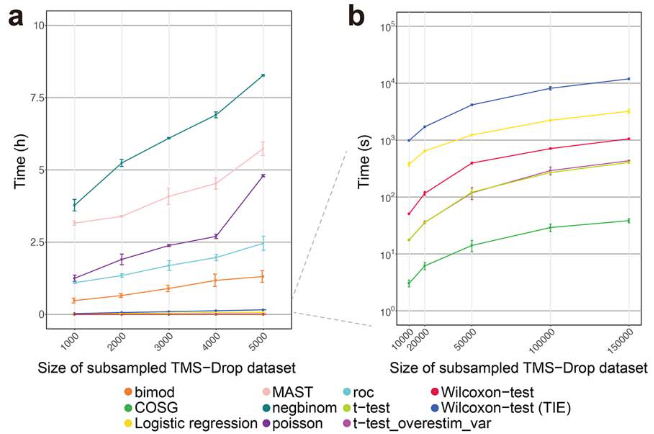
(2)在大规模单细胞数据中, COSG在保持精度的同时分析速度很快;
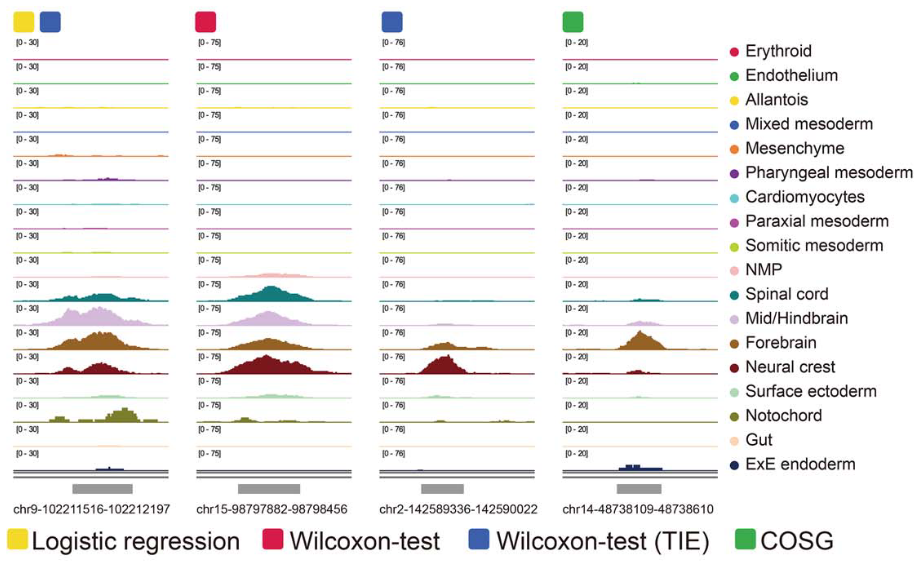
(3)COSG在scATAC以及空间转录组数据上也同样具有较优的表现
代码实操
remotes::install_github(repo = 'genecell/COSGR')
library(COSG)
library(Seurat)
## 输入数据
data('pbmc_small',package='COSG')
dim(pbmc_small@assays$RNA@data) #标准化矩阵
# [1] 230 80
table(Idents(pbmc_small)) #聚类分群结果
# 0 1 2
# 36 25 19 - R包的分析函数即为
cosg(),默认参数如下
marker_cosg <- cosg(
pbmc_small,
groups='all', #考虑全部分组
assay='RNA',
slot='data',
mu=1, #惩罚项参数,值越大
remove_lowly_expressed=TRUE, #是否过滤低表达基因
expressed_pct=0.1, #设置低表达的阈值
n_genes_user=100 #每个cluster定义Top-N个marker gene
)
head(marker_cosg$names,3)
# 0 1 2
# 1 CD7 S100A8 MS4A1
# 2 CCL5 TYMP CD79A
# 3 GNLY S100A9 TCL1A
head(marker_cosg$scores,3)
# 0 1 2
# 1 0.6391917 0.8954042 0.6922908
# 2 0.6391267 0.8312083 0.5832425
# 3 0.6328148 0.8120045 0.5757478- 简单可视化一下
top_list<-c()
for (group in colnames(marker_cosg$names)){
top_i<-marker_cosg$names[group][1:5,1]
top_list<-c(top_list,top_i)
}
DotPlot(pbmc_small,
assay = 'RNA',
# scale=TRUE,
features = unique(top_list)) + RotatedAxis()
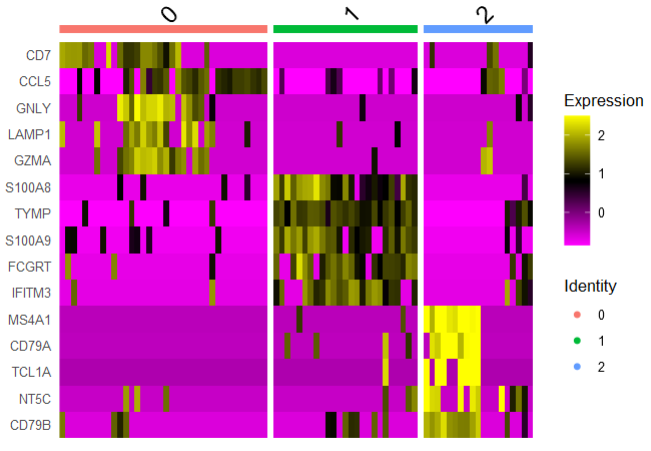
pbmc_small <- ScaleData(pbmc_small, features = top_list)
DoHeatmap(pbmc_small,
assay = 'RNA',
features = top_list) 
tips
Note
正常NFS推荐使用该方法,作者不推荐在SCTransform之后使用该方法
The above error might be caused by using the SCT assay, which is not sparse after the SCTransform normalization. We recommend use the
RNAassay with the slotdataas the input.