论文
主要观点:针对Bulk数据开发的特征评分方法(如ssGSEA)不建议用于癌症单细胞 RNA 测序数据
- Noureen N, Ye Z, Chen Y, Wang X, Zheng S. Signature-scoring methods developed for bulk samples are not adequate for cancer single-cell RNA sequencing data. Robles-Espinoza CD, Barkai N, Han L, eds. eLife. 2022;11:e71994. doi:10.7554/eLife.71994
研究背景
-
研究问题:这篇文章研究了在单细胞RNA测序(scRNA-seq)数据分析中,批量样本的基因表达签名评分方法是否适用于癌症单细胞数据。具体来说,文章探讨了批量样本方法(如ssGSEA和GSVA)在癌症单细胞数据中的表现,并提出需要谨慎使用这些方法。
-
研究难点:该问题的研究难点包括:scRNA-seq数据的高丢失率(dropout rates),这可能导致非表达基因在基因签名中的不平衡表示,从而引入评分偏差;癌细胞通常表达更多的基因,这种不平衡会影响基于批量样本的方法的表现。
-
相关工作:该问题的研究相关工作包括:Holland等人(2020)和张等人(2020)对某些应用的基准测试,以及其他一些单细胞RNA测序数据分析方法的开发和应用。
研究方法
这篇论文提出了五种签名评分方法,用于解决批量样本方法在单细胞RNA测序数据中的适用性问题。具体来说,
- 单样本基因集富集分析(ssGSEA):该方法通过计算基因集在样本中的富集程度来评分基因签名。其计算公式如下:
$$\text{ssGSEA score} = \sum_{i=1}^{m} R_{g, i}/(m\times N)$$
-
基因集变异分析(GSVA):该方法通过计算基因集在样本中的变异程度来评分基因签名。其计算过程与ssGSEA类似,但使用了不同的统计量。
-
AUCell:该方法使用曲线下面积(AUC)来测试基因集在细胞中前N个高表达基因中的富集程度。
-
单细胞签名探索器(SCSE):该方法通过归一化签名基因的总表达量来评分基因签名。
-
联合评估签名均值和推断富集(JASMINE):该方法结合了签名基因的排名和富集程度来评分基因签名。其计算公式如下:
$$V_ {\text{mean}}=\sum_{i=1}^{m} R_{g, i}/(m\times N)$$
$$OR=(a* d)/(b* c)$$
其中,Rg 表示基因 g 在所有表达基因中的排名,a 表示签名基因中表达的基因数量,b 表示签名基因中未表达的基因数量,c 表示非签名基因中表达的基因数量,d 表示非签名基因中未表达的基因数量。
实验设计
-
数据收集:收集了10个不同癌症类型的单细胞RNA测序数据集,并从原始研究中获取了细胞类型注释。
-
数据预处理:过滤掉非表达基因,并使用Seurat中的正则化负二项回归进行归一化。
-
基因集选择:从MSigDB中选择了7503个至少有20个基因的基因集。
-
方法测试:使用ssGSEA、GSVA、AUCell、SCSE和JASMINE五种方法对基因集进行评分,并比较其在肿瘤和正常细胞中的表现。
结果与分析
- 基因计数不平衡:在所有数据集中,肿瘤细胞的平均检测基因数量显著高于正常细胞。即使在将正常细胞分为不同细胞群体的情况下,这一不平衡仍然存在。
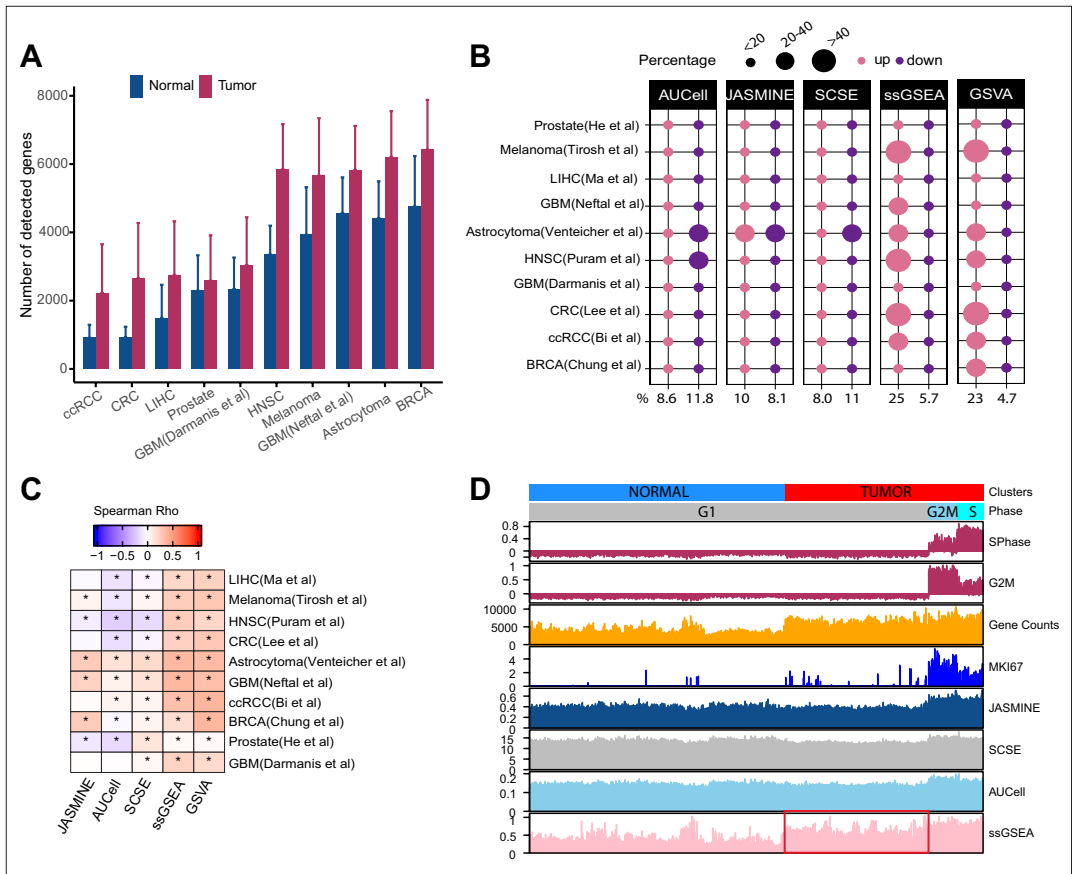
-
ssGSEA签名评分的偏差:ssGSEA和GSVA在高基因计数的细胞类型中,如TEC、TAM和CAF,未能显著识别出更多的上调基因集。此外,ssGSEA和GSVA的Cohen's d值与基因集大小呈正相关,而单细胞方法则没有这种模式。
-
敏感性和特异性:在模拟真实世界的噪声水平下,所有方法在80%噪声水平下仍能检测到50%的上调基因集,但ssGSEA在下调基因集的检测上表现较差。在空采样实验中,AUCell和JASMINE在特异性方面表现优于SCSE和ssGSEA。
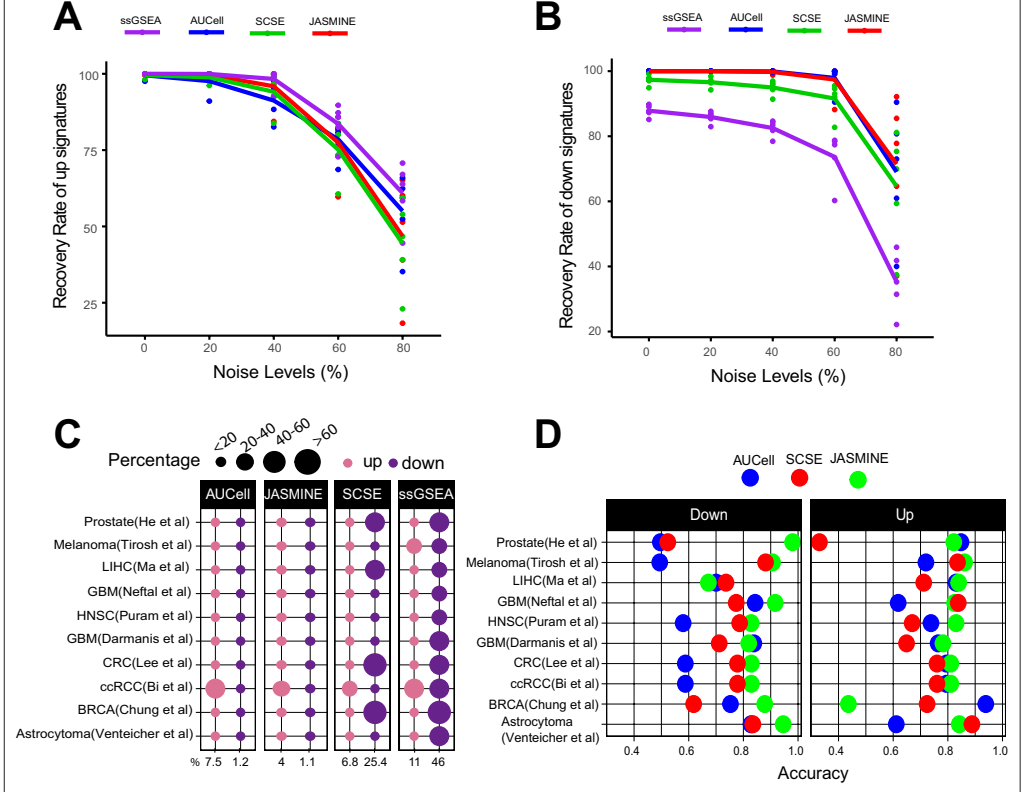
-
与共识的一致性:JASMINE在大多数数据集中与共识基因集的一致性较好,而AUCell在下调基因集的准确性较低。
-
计算效率:SCSE在计算效率上表现最佳,而JASMINE虽然速度较快,但内存需求较高。
总体结论
这篇论文通过对五种签名评分方法的基准测试,发现基于批量样本的方法在单细胞RNA测序数据中存在显著的偏差,特别是在癌细胞中。单细胞方法相对更稳健,能够更好地识别真实的上调和下调基因签名。研究结果表明,在使用批量样本方法时应谨慎,并在设计基准测试策略时考虑细胞上下文。
论文评价
优点与创新
-
全面的基准测试:论文对五种签名评分方法进行了全面的基准测试,包括单样本基因集富集分析(ssGSEA)、基因集变异分析(GSVA)、AUCell、单细胞签名探索器(SCSE)和一种新方法——联合评估签名均值和推断富集(JASMINE)。
-
癌症数据集的验证:使用癌症作为示例,展示了癌症细胞相比正常细胞表达更多基因的现象,并证明了这种不平衡会导致基于批量样本的ssGSEA在金标准测试和下采样实验中的偏差。
-
新方法的开发:提出了一种新方法JASMINE,该方法通过考虑表达基因中的签名基因的富集和平均表达水平来评估签名,从而减少dropout的影响。
-
对dropout的深入分析:详细探讨了dropout对ssGSEA评分的影响,并通过模拟实验展示了dropout如何改变签名评分。
-
敏感性、特异性和准确性的比较:对四种单细胞方法和两种批量样本方法在不同噪声水平和下采样率下的检测敏感性和特异性进行了详细的比较。
不足与反思
-
GSVA的速度问题:由于GSVA的运行速度较慢,且其输出与ssGSEA高度相关,因此在后续的金标准测试和下采样实验中将其移除。
-
批量样本方法的局限性:论文指出,批量样本方法如ssGSEA和GSVA未设计处理过多的dropout,这在细胞生物学背景下可能导致误导性甚至错误的数据分析。
-
未来工作的方向:建议在未来的研究中考虑细胞生物学背景下的dropout rate差异,并在基准测试策略中加以考虑。